Projektgruppe: Sebastian Herbig, Essen, Vanessa Kaiser, Mainz, Jürgen Maurer, Wetzlar, Lenka Taylor, Heidelberg, Judith Thiesen, Mainz, Irene Krämer, Mainz (Leitung)
1. Zweckbestimmung und Geltungsbereich
Diese Leitlinie beschreibt die Verfahrensweise zur aseptischen Herstellung und Prüfung applikationsfertiger Parenteralia ohne Anwendung eines Sterilisationsverfahrens (Sterilfiltration, Sterilisation im Endbehältnis) in der Krankenhausapotheke. Diese findet im Rahmen des üblichen Apothekenbetriebs statt und dient der Versorgung von Bereichen, in denen eine qualitätsgesicherte Zubereitung unter aseptischen Bedingungen nicht hinreichend möglich ist. Es handelt sich bei der Herstellung um eine aseptische Einzelherstellung von Parenteralia in patientenindividueller Dosierung (Rezeptur, z. B. Zytostatikazubereitungen, Mischinfusionslösungen für die total parenterale Ernährung) oder um die aseptische Herstellung applikationsfertiger Parenteralia in Standarddosierungen (Defektur, z. B. Zytostatika in Standarddosierung für intraokulare Injektion, Standardmischinfusionslösungen für die total parenterale Ernährung, Lösungen für die Epiduralanalgesie, sonstige (Dauer)injektionen, -infusionen für Intensivpatienten). Die Produkte werden in der Regel unverändert direkt aus dem Primärbehältnis appliziert. Bei der Herstellung werden sterile Ausgangsprodukte (Fertigarzneimittel, Medizinprodukte, defekturmäßig eigenhergestellte Parenteralia) verwendet. Die Herstellung wird in möglichst wenigen aseptischen Herstellungsschritten durchgeführt und unverzüglich abgeschlossen. Die hergestellten applikationsfertigen Parenteralia sind zum alsbaldigen Verbrauch bestimmt. Lagerbedingungen, Haltbarkeit und Aufbrauchfrist sollen auf Grundlage der physikalisch-chemischen Stabilität, mikrobiologischen Validierung und einer Risikobewertung festgelegt werden.
2. Regulatorische Anforderungen
Die Herstellung von Arzneimitteln zur parenteralen Anwendung muss im Qualitätsmanagementsystem der Apotheke festgelegt sein. Die Sicherstellung der ordnungsgemäßen Qualität von in der Apotheke aseptisch hergestellten applikationsfertigen Parenteralia erfolgt unter Beachtung der allgemein anerkannten pharmazeutischen Regeln. Bei der Erstellung dieser Leitlinie wurden insbesondere berücksichtigt:
- Apothekenbetriebsordnung §§ 2a, 3, 4, 4a, 6, 7, 8, 14, 35
- PIC/S PE 010-3 Good Preparation Practice (GPP), Guide to good practices for the preparation of medicinal products in healthcare establishments
- Ph. Eur. 7.4 (incl. Draft Monograph Pharmaceutical Preparations, Praeparationes Pharmaceuticae)
- Council of Europe Resolution CM/ResAP (2011)1
- USP 35/NF 30, Monographie <797> Pharmaceutical Compounding – Sterile Preparations
Bei der Herstellung von CMR-Arzneimitteln sind die geltenden Vorschriften zum Arbeitsschutz (Arbeitsstättenverordnung, TRGS 525, BGW Themenheft Zytostatika im Gesundheitsdienst) zu berücksichtigen.
Die Unterscheidung von Rezeptur- und Defekturarzneimitteln erfolgt entsprechend der Apothekenbetriebsordnung.
3. Zuständigkeiten
Die Herstellung und Prüfung von Arzneimitteln ist gemäß § 1a Abs. 3 ApBetrO eine pharmazeutische Tätigkeit und wird vom pharmazeutischen Personal gemäß § 1a Abs. 2 ApBetrO durchgeführt. Die aseptische Herstellung applikationsfertiger Parenteralia darf gemäß § 35 ApBetrO nur von geschultem und qualifiziertem Personal vorgenommen werden. Für die ordnungsgemäße Plausibilitätsprüfung, Herstellung, Prüfung, Freigabe und Abgabe tragen der zuständige Apotheker sowie der Apothekenleiter die Verantwortung.
Nichtpharmazeutisches Personal darf gemäß § 3 Abs. 5a ApBetrO das pharmazeutische Personal im Rahmen der pharmazeutischen Tätigkeiten unterstützen.
4. Qualitätsstandard für die aseptische Herstellung und Prüfung applikationsfertiger Parenteralia
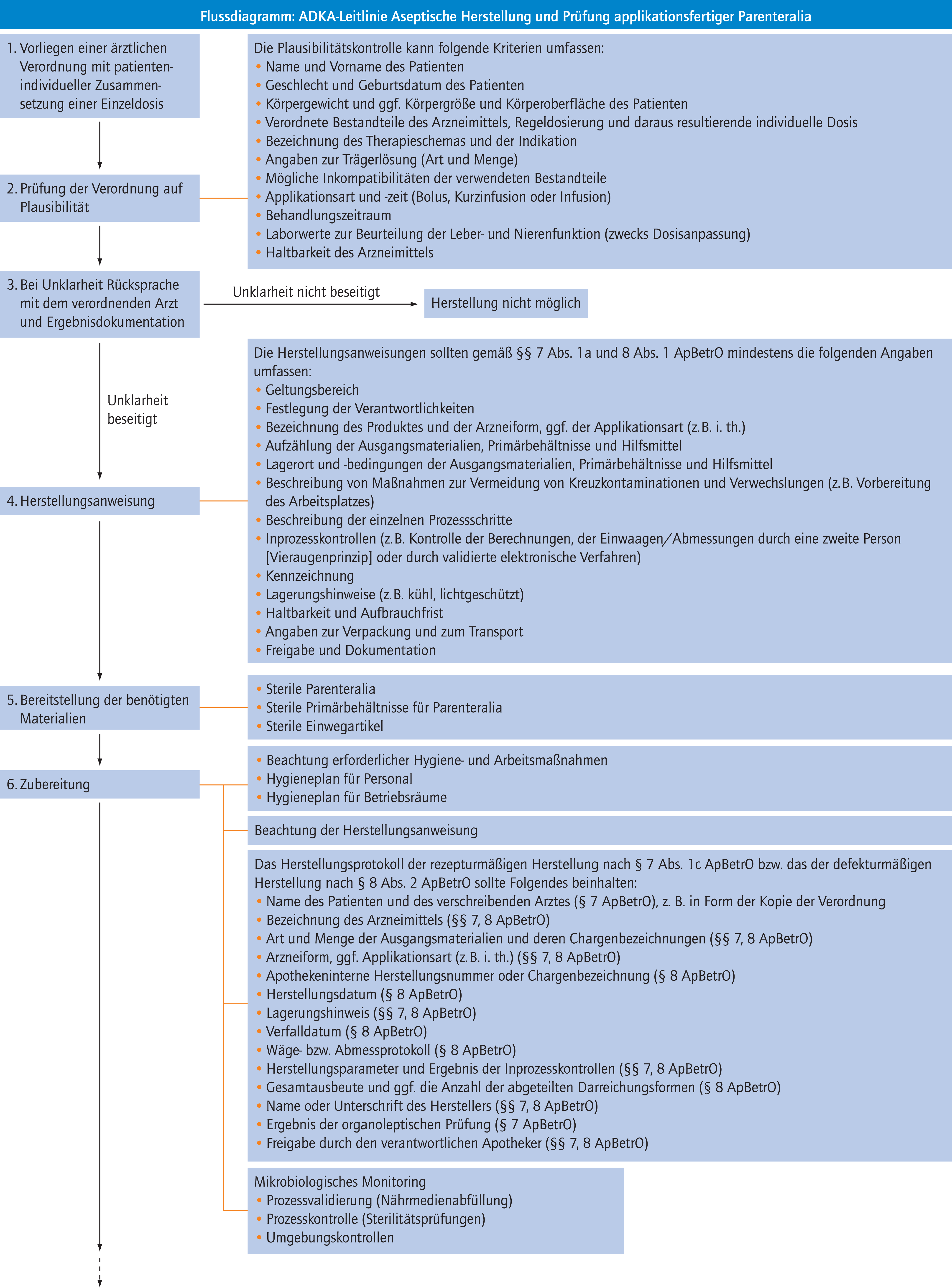
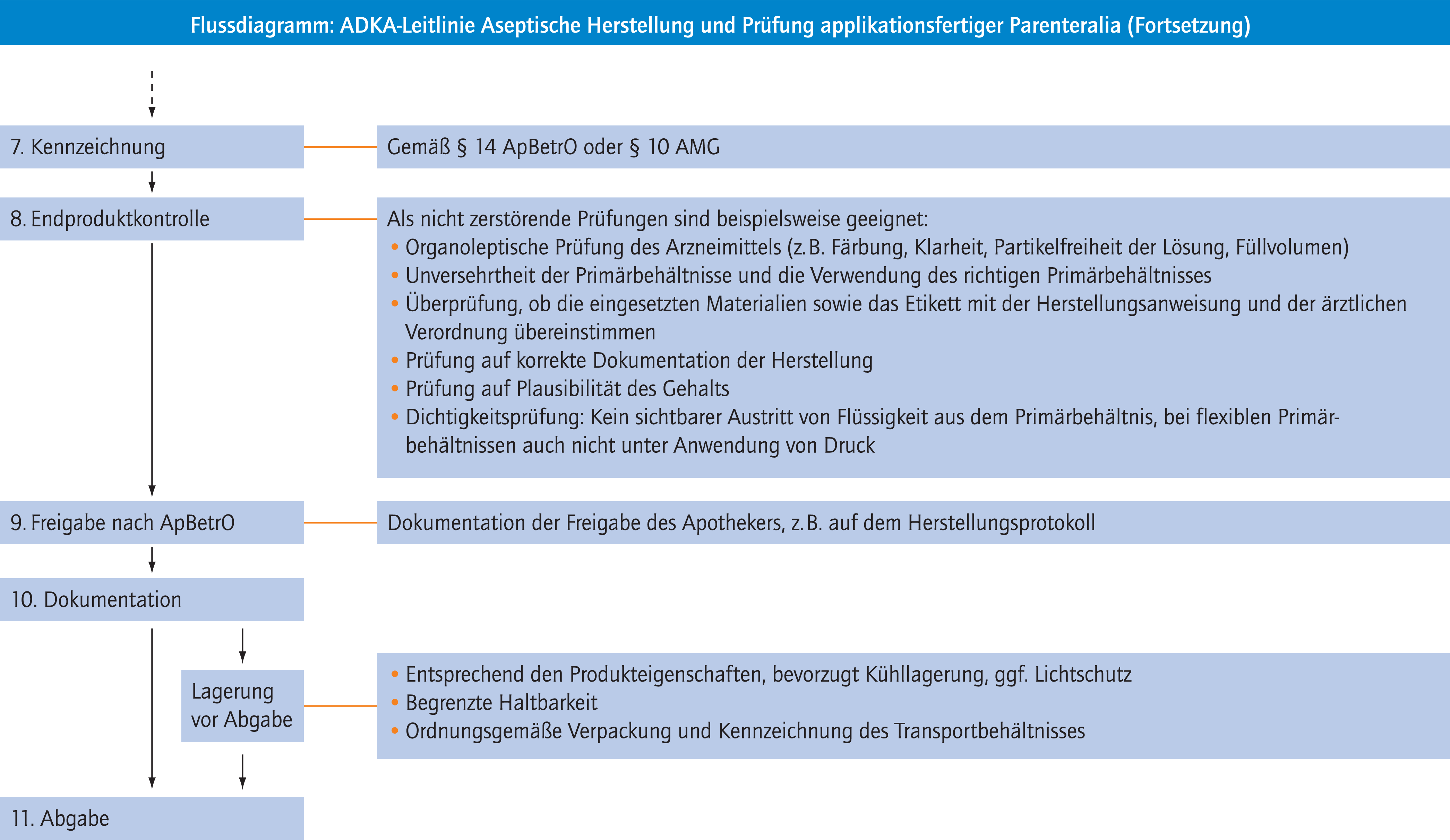
Siehe Flussdiagramm.
Anhang
I-1 Plausibilitätskontrolle der Verordnung
Die Anforderung für die Herstellung eines Rezepturarzneimittels ist von einem Apotheker einer Plausibilitätskontrolle gemäß § 7 Abs. 1b und § 35 Abs. 6 ApBetrO zu unterziehen. Unklarheiten sind gemäß § 7 Abs. 1 ApBetrO durch Rücksprache mit dem verordnenden Arzt zu beseitigen. Die Plausibilitätskontrolle kann folgende Kriterien umfassen:
- Name und Vorname des Patienten
- Geschlecht und Geburtsdatum des Patienten
- Körpergewicht und ggf. Körpergröße und Körperoberfläche des Patienten
- Verordnete Bestandteile des Arzneimittels, Regeldosierung und daraus resultierende individuelle Dosis
- Bezeichnung des Therapieschemas und der Indikation
- Angaben zur Trägerlösung (Art und Menge)
- Mögliche Inkompatibilitäten der verwendeten Bestandteile
- Applikationsart (Bolus, Kurzinfusion oder Infusion)
- Behandlungszeitraum
- Laborwerte zur Beurteilung der Leber- und Nierenfunktion (zwecks Dosisanpassung)
- Haltbarkeit des Arzneimittels
Die Plausibilitätsprüfung ist gemäß § 7 Abs. 1b ApBetrO von einem Apotheker zu dokumentieren (siehe Anweisung im Flussdiagramm und Appendix 1, 2 beispielhafte Dokumentation der Plausibilitätsprüfung). Bei elektronischer Verordnung kann die Plausibilitätskontrolle und deren Dokumentation entsprechend angepasst werden. Die Dokumentation ist mit Datum und Namenszeichen des Apothekers zu versehen.
Appendix 2, Beispiel 1. Plausibilitätsprüfung der Verordnung mittels Checkliste. Die Plausibilitätsprüfung der Zytostatikaverordnung sollte gemäß § 7 Abs. 1b sowie § 35 Abs. 6 ApBetrO und gemäß BAK-Leitlinie zur aseptischen Herstellung und Prüfung applikationsfertiger Parenteralia mit CMR-Eigenschaften folgende Punkte umfassen:
|
Station |
Zwecks Belastung der Kostenstelle, Anlieferadresse, Rückfragen |
|
Name, Vorname, Geschlecht |
Identifikation des Patienten |
|
Geburtsdatum |
Eindeutige Identifikation des Patienten und Abschätzung altersbedingter Dosismodifikationen |
|
Köpergewicht, ggf. Körpergröße, ggf. Körperoberfläche |
Zur Kontrolle der Körperoberfläche und der Dosisberechnung |
|
Patientenindividuelle Faktoren, wie Leber- und Nierenwerte |
Zur Abschätzung nötiger Dosismodifikationen bzw. Carboplatindosierungen |
|
Diagnose |
Zur Überprüfung, ob das angegebene Chemotherapieprotokoll plausibel ist |
|
Name und Zusammensetzung des Chemotherapieprotokolls |
Identifikation des Protokolls und Vergleich mit dem Originalprotokoll |
|
Ggf. Studienbezeichnung, ZE/NUB-Bezeichnung |
|
|
Angaben zur Dosismodifikation |
|
|
Applikationsdatum |
|
|
Therapietag im Protokoll |
|
|
Art und Menge der Arzneimittel/Wirkstoffe |
|
|
Regeldosierung und errechnete Dosis |
Nachvollziehen der Dosisberechnung |
|
Art und Volumen der Trägerlösung |
|
|
Häufigkeit und Dauer der Infusion |
|
|
Applikationsart, Applikationssystem, Applikationszeit |
|
|
Inkompatibilitäten, Instabilitäten, Interaktionen |
|
|
Haltbarkeit |
|
|
Arztunterschrift |
ZE: Zusatzentgelt; NUB: Neue Untersuchungs- und Behandlungsmethoden
I-2 Ausgangsmaterialien und Betriebsmittel
I-2.1 Ausgangsmaterialien
Als Ausgangsmaterialien werden ausschließlich für die parenterale Anwendung bestimmte Fertigarzneimittel, defekturmäßig eigenhergestellte Parenteralia, Medizinprodukte und sterile Behältnisse verwendet, deren pharmazeutische Qualität sichergestellt ist.
Spezifikation der eingesetzten Parenteralia
- Fertigarzneimittel und defekturmäßig eigenhergestellte Parenteralia sind in Verpackungen zu verwenden, die helfen, die mikrobiologische Ausgangsbelastung im Herstellungsbereich gering zu halten.
- Sind Ausgangsmaterialien nicht steril verpackt, sind geeignete Desinfektionsmaßnahmen durchzuführen (siehe I-5.3).
Spezifikation der eingesetzten Primärbehältnisse
- Es werden sterile Behältnisse für Parenteralia eingesetzt. In der Regel werden leere oder mit Trägerlösung vorgefüllte Behältnisse verwendet, zum Beispiel Einmalspritzen, Infusionsbeutel, Reservoirs für tragbare Pumpen.
- Die pharmazeutische Qualität der Behältnisse muss durch ein Prüfzertifikat des Herstellers belegt sein. Soweit es sich um Medizinprodukte handelt, ist die Konformität mit dem Medizinproduktegesetz (i. e. CE-Kennzeichnung) sicherzustellen.
- Die Primärbehältnisse sind so zu wählen, dass Inkompatibilitäten nicht zu erwarten sind.
- Die Dichtigkeit der Behältnisse nach Befüllung muss in geeigneter Weise sichergestellt werden, zum Beispiel Kombiverschluss bei Einmalspritzen, bei Infusionsbeuteln elastischer Stopfen, der sich wiederverschließt.
Qualitätsprüfung der Ausgangsmaterialien
Die eingesetzten Fertigarzneimittel und Primärbehältnisse werden regelmäßig gemäß § 12 ApBetrO stichprobenweise geprüft. Bei zerstörenden Prüfungen vor der Herstellung sind die betreffenden Packungen von der aseptischen Herstellung auszuschließen. Alle Untersuchungsergebnisse werden dokumentiert (Gestaltung des Prüfprotokolls siehe Appendix 3, Bsp. 2, beispielhaftes Formblatt [Appendix 3 Bsp. 1 ist zu finden in Krankenhauspharmazie 2003;24:201]).
Die Prüfung der Ausgangsmaterialien sollte mindestens folgende Kriterien umfassen:
- Farbe
- Klarheit
- Unversehrtheit der Primärbehältnisse und ggf. der Sterilschutzumhüllungen
- Haltbarkeitsdatum
- Freiheit von Partikeln (Ph. Eur. Partikelkontamination – sichtbare Partikel) und sonstigen Verunreinigungen
- Füllvolumen
Die Prüfung der sterilen Primärbehältnisse sollte mindestens folgende Kriterien umfassen:
- Unversehrtheit der Verpackung
- Freiheit von Partikeln und sonstigen Verunreinigungen
- Haltbarkeitsdatum
I-2.2 Hilfsmittel
Bei der aseptischen Herstellung werden sterile Einwegartikel verwendet, zum Beispiel Einmalspritzen und Kanülen, Spikes, Adapter, Überleitsysteme und Verschlusskonen. Soweit es sich um Medizinprodukte handelt, ist die Konformität mit dem Medizinproduktegesetz (i. e. CE-Kennzeichnung) sicherzustellen.
I-2.3 Reinigungs- und Desinfektionsmittel
- Für die Reinigung und Desinfektion der Räume, Werkbänke und Materialien sind geeignete Reinigungs- und Desinfektionsmittel gemäß VAH-Liste und – falls zutreffend – deren einzusetzende Verdünnung im Hygieneplan festzulegen. Die Auswahl der Reinigungs- und Desinfektionsmittel sollte sich am Standard des jeweiligen Krankenhauses mit den Konzentrationen und Einwirkzeiten für Risikobereiche orientieren. In begründeten Fällen (z. B. Anstieg der Keimbelastung) sind sporozide Desinfektionsmittel zu verwenden.
- Desinfektionsmittel zum Einsatz in Reinraumklasse A und B müssen sporenfrei sein.
- Aufbrauch- bzw. Standfristen für Reinigungs- und Desinfektionsmittel müssen im Hygieneplan festgelegt werden.
I-2.4 Betriebsmittel
Es sind Festlegungen zur Qualifizierung, Kalibrierung, Wartung und Reinigung der Betriebsmittel zu treffen.
Zur Unterstützung der Herstellung applikationsfertiger Parenteralia empfiehlt sich die Verwendung einer geeigneten Software mit geeigneten Funktionalitäten für Plausibilitätskontrollen, Dokumentenerstellung und die Dokumentation.
I-3 Räumlichkeiten
Anforderungen an die Herstellungsräumlichkeiten
- Der Herstellungsraum muss gemäß § 35 Abs. 3 ApBetrO ausschließlich dem Zweck der Herstellung parenteraler oder anderer steriler Zubereitungen gemäß Arzneibuch dienen. Bei Herstellung unterschiedlicher Produktgruppen im gleichen Raum (CMR-Arzneimittel, Nicht-CMR-Arzneimittel) muss die Kreuzkontamination durch organisatorische und Reinigungsmaßnahmen ausgeschlossen sein.
- Der Zugang muss über einen Zwischenraum (Schleuse) erfolgen, der für die Aufrechterhaltung der im Herstellungsraum erforderlichen Reinraumklassen geeignet ist.
- Die Belüftung muss über Filter angemessener Wirksamkeit (z. B. HEPA-Filter Klasse H13, H14) erfolgen.
- Die Druckunterschiede sollen dem Zonenkonzept entsprechend eingerichtet (10–15 Pa Differenz) und überwacht werden; Orientierungszahlen für Luftwechselzahlen: Reinraumklasse D 10-fach; C 30-fach; B 40-fach.
- Fenster sind zu jeder Zeit, Türen sind während der Herstellung geschlossen zu halten.
- Wände, Decken, Fußböden und Arbeitsflächen sollten glatte, fugenlose Oberflächen haben und gut zu reinigen beziehungsweise zu desinfizieren sein.
- Fußbodenbeläge sollten fugenlos sein.
- Waschbecken und Abflüsse dürfen (Reinraumklasse B) bzw. sollen (Reinraumklasse C, D) im Herstellungsraum nicht vorhanden sein.
- Die Ausstattung mit Mobiliar und Geräten soll auf das Notwendige beschränkt sein.
- In den Herstellungsräumen dürfen sich nur Mitarbeiter aufhalten, die dort entsprechende Tätigkeiten ausführen. Während der Herstellung ist die Zahl der anwesenden Mitarbeiter auf ein Minimum zu reduzieren.
I-3.1 Reinraumqualität
Alle kritischen Arbeitsschritte müssen unter Laminar-Airflow-(LAF-)Bedingungen durchgeführt werden. Es kommen bevorzugt Vertikal-LAF-Werkbänke zum Einsatz.
Durch entsprechende Validierung ist nachzuweisen, dass im Arbeitsbereich während der Zubereitung und Abfüllung ein Luftreinheitsgrad für Keimzahl und Partikelzahl entsprechend der Klasse A des EG-GMP Leitfaden Anhang 1 gegeben ist (siehe Appendix 4, Tabelle Reinraumklassen). Der kontrollierte Bereich kann abweichend von Klasse B mindestens der Klasse C des Anhangs des Leitfadens entsprechen, wenn die Arzneimittelqualität durch das angewendete Verfahren nachweislich gewährleistet wird und durch entsprechende Validierung des Verfahrens belegt ist (s. Punkt I-10). Bei Einsatz eines Isolators soll der kontrollierte Bereich der Klasse D des EG-GMP Leitfadens Annex 1 entsprechen.
Appendix 4. Reinraumklassen gemäß EU-GMP-Guide. Partikelmesswerte
|
Maximal erlaubte Partikelzahl pro m3 (gleich oder größer als die aufgeführte Größe) |
||||
|
Ruhezustand |
Betriebszustand |
|||
|
Klasse |
0,5 µm |
5,0 µm |
0,5 µm |
5,0 µm |
|
A |
3520 |
20 |
3520 |
20 |
|
B |
3520 |
29 |
352000 |
2900 |
|
C |
352000 |
2900 |
3520000 |
29000 |
|
D |
3520000 |
29000 |
Nicht festgelegt |
Nicht festgelegt |
Appendix 4. Reinraumklassen gemäß EU-GMP-Guide. Keimzahlen im Betriebszustand
|
Empfohlene Grenzwerte für die mikrobiologische Kontaminationa |
||||
|
Klasse |
Luftprobe [KBE/m3] |
Sedimentationsplatten [KBE/4 Stundenb] |
Kontaktplatten [KBE/Platte] |
Handschuhabdruck 5 Finger [KBE/Handschuh] |
|
A |
< 1 |
< 1 |
< 1 |
< 1 |
|
B |
10 |
5 |
5 |
5 |
|
C |
100 |
50 |
25 |
– |
|
D |
200 |
100 |
50 |
– |
a: Hierbei handelt es sich um Durchschnittswerte; b: Einzelne Sedimentationsplatten können weniger als 4 Stunden exponiert werden.
Die Wartung und Klassifizierung der Reinraumbereiche sollte jährlich entsprechend der DIN EN ISO 14644 erfolgen (Partikelzahl, Keimzahl, Luftwechselzahl, Filterintegrität).
I-3.2 Raum mit Schleusenfunktion
Der Zugang in den kontrollierten Bereich muss gemäß § 35 Abs. 3 ApBetrO über einen Raum mit Schleusenfunktion und Waschgelegenheit erfolgen. Im Schleusenbereich ist die spezielle Bereichskleidung anzulegen. Wenn möglich, sollte Material getrennt vom Personal eingeschleust werden. Es muss technisch oder organisatorisch sichergestellt sein, dass die Türen einer Schleuse nicht gleichzeitig geöffnet werden.
I-3.3 Laminar-Airflow-(LAF-)Werkbänke, Isolatoren
Die LAF-Werkbänke (nach DIN 12980, DIN EN 12469) und Isolatoren sind sachgerecht aufzustellen, zu betreiben, zu prüfen und zu warten. Vor Erstinbetriebnahme sowie nach jedem Filterwechsel oder sonstigen Veränderungen des Aufstellungsortes und nach jeder baulichen Veränderung, jedoch mindestens einmal jährlich, muss die Funktionsfähigkeit durch Sachkundige überprüft werden.
LAF-Werkbänke sollten vorzugsweise kontinuierlich in Betrieb sein. Ist dies nicht der Fall, muss eine ausreichende Vorlaufzeit (entsprechend der Bedienungsanleitung) gewährt werden, um die erforderlichen Reinheitsbedingungen zu erreichen.
Beim Umgang mit CMR-Arzneimitteln muss eine Sicherheitswerkbank (bevorzugt nach DIN 12980) eingesetzt werden.
Bei Isolatoren ist wöchentlich ein Druckhaltetest durchzuführen und bei jedem Arbeitsbeginn die Unversehrtheit der Handschuhe visuell zu prüfen.
I-4 Personal
I-4.1 Personal und Arbeitsweise
Die aseptische Herstellung darf nur von geschultem und mittels Nährmedienabfüllung geprüftem pharmazeutischen Personal durchgeführt werden. Der Ersteinweisung von Mitarbeitern muss größte Aufmerksamkeit gewidmet werden, beispielsweise durch praktische Übungen an applikationsfertigen Parenteralia, die nicht für die Anwendung bestimmt sind. Die Qualifikation der Mitarbeiter, einschließlich Reinigungspersonal, muss in regelmäßigen Abständen (mindestens einmal jährlich) durch interne und/oder externe Schulungen gesichert werden (Schulungsinhalte: Personalhygiene, Raumhygiene, Verhalten im Reinraum, aseptische Arbeitsweisen, Umgebungsmonitoring, sicherer Umgang mit CMR-Arzneimitteln, Dokumentation). Die Schulungsmaßnahmen sind zu dokumentieren. Die Teilnahme an der Schulung ist von den Mitarbeitern zu bestätigen. Der verantwortliche Apotheker kann entscheiden, ob und in welcher Art eine Erfolgskontrolle durchgeführt wird.
I-5 Hygienemaßnahmen
Bei allen Herstellungsprozessen müssen gemäß § 4a ApBetrO geeignete Hygienemaßnahmen getroffen werden, die die mikrobiologische Qualität des Arzneimittels sicherstellen. Die erforderlichen Hygienemaßnahmen für Personal, Betriebsräume und Materialien müssen schriftlich in einem Hygieneplan festgelegt werden. Die Durchführung der Raumhygiene muss pro Reinigungsmaßnahme dokumentiert werden (siehe Appendix 5, beispielhafte Dokumentationsformulare).
I-5.1 Hygieneplan
Der Hygieneplan sollte enthalten:
- Geltungsbereich
- Festlegung der Verantwortlichkeit (u. a. Benennung eines Hygienebeauftragten)
- Einteilung der Arbeitsbereiche für unterschiedliche Herstellungsschritte in unterschiedliche Reinraumzonen, zum Beispiel Zubereitung, Kennzeichnung, Dokumentation
- Zutrittsregelungen
- Personalhygiene mit Einstufung der Schutz- und Reinraumkleidung unter Beachtung der entsprechenden Tätigkeiten und Aufenthalt in den verschiedenen Reinraumzonen
- Raumhygiene (inkl. Reinigungsplan)
- Ein- und Ausschleusen von Materialien und Personal (siehe Appendix 6, beispielhafte Anleitung)
- Liste der verwendeten Reinigungs- und Desinfektionsmittel und Arbeitsgeräte
- Dokumentation der Reinigungsmaßnahmen
I-5.2 Raumhygiene
Alle Betriebsräume für die aseptische Herstellung sind nach schriftlichen Reinigungs- und Desinfektionsplänen (siehe Appendix 7, beispielhafte Anleitungen) regelmäßig zu reinigen. Die kritischen Bereiche (LAF) sollten von Personal gereinigt werden, das im Herstellungsbereich tätig ist oder speziell dafür eingearbeitet wurde. Im Hygieneplan müssen mindestens Festlegungen getroffen werden zu:
- Art und Häufigkeit der Reinigung/Desinfektion
- einzusetzenden Reinigungs- und Desinfektionsmitteln und falls zutreffend deren einzusetzende Verdünnung und Einwirkzeit
- einzusetzenden Arbeitsgeräten (Keim- und fusselarme Wischkörper)
- Zutrittsregelung für Herstellungsbereiche.
I-5.3 Personalhygiene
- Für die Personalhygiene ist eine Anweisung zu erstellen, in die die Mitarbeiter einzuweisen und regelmäßig zu unterweisen sind (s. Punkt I-4).
- Schleusen zu Reinraumbereichen C müssen mindestens mit apothekenüblicher Bereichskleidung betreten werden.
- Das Personal hat im Herstellungsbereich mindestens sterilisierte oder desinfizierend gewaschene Bereichskleidung mit langen Ärmeln und eng anliegenden Bündchen (z. B. hinten geschlossene Kittel oder Overall), Strümpfe sowie desinfizierbare Schuhe zu tragen (siehe Appendix 8, Abbildung zu Bekleidungsregeln).
- Im Herstellungsraum sind immer Haube, Mundschutz, ggf. Bartbinde und Handschuhe zu tragen. Haar und ggf. Bart bzw. Schnurrbart sind vollständig abzudecken.
- Beim Arbeiten an der Werkbank sind sterile Handschuhe anzulegen und regelmäßig zu wechseln.
- Händedesinfektion: Die Hände incl. Handgelenke und ggf. Unterarme sollten zunächst gewaschen werden. Die trockenen Hände und ggf. Unterarme sind anschließend einer Desinfektion (Einwirkzeit mindestens 30 s) gemäß hausinternem Standard zu unterziehen.
- Die Bereichskleidung muss täglich gewechselt werden.
- Kopfhaube und Mundschutz sollten nach jeder Arbeitsperiode gewechselt werden.
- Handschuhe sind regelmäßig zu desinfizieren (außer beim Umgang mit CMR-Arzneimitteln) und zu wechseln (z. B. alle 30 Minuten). Bei jedem Handschuhwechsel ist eine Händedesinfektion vor dem Anlegen der neuen Handschuhe durchzuführen.
- Beim Umgang mit CMR-Arzneimitteln sind Personenschutzmaßnahmen einzuhalten und ist die persönliche Schutzausrüstung (s. TRGS 525, BGW Themenheft „Zytostatika im Gesundheitsdienst“) zu tragen.
Appendix 8. Beispiele für Bekleidungsregeln. Links: Beispiel für apothekenübliche Bereichskleidung; rechts: Beispiel für Reinraumkleidung für Reinraumklasse B und C

I-5.4 Umgang mit Ausgangsmaterialien
- Die Ausgangsmaterialien sind ohne Kartonagen in den Herstellungsraum einzuschleusen.
- Sind die Ausgangsmaterialien nicht steril verpackt, sind sie vor Einbringen in den Herstellungsraum bzw. in die LAF-Werkbank gemäß einer im Hygieneplan festgelegten (Wisch-)Desinfektion zu desinfizieren (Ausnahme: einfachverpackte Sterilgüter).
- Besondere Sorgfalt ist im Rahmen der Zubereitung auf die Desinfektion der Anstichstellen zu legen.
- Bei CMR-Fertigarzneimitteln sind die Desinfektionsmaßnahmen so durchzuführen, dass der Personenschutz gewährleistet ist (keine offene Sprühdesinfektion).
I-6 Arbeitsschutzmaßnahmen
Im Umgang mit stark wirksamen Arzneistoffen, die als Gefahrstoffe gemäß GefStoffV eingeordnet werden (§§ 2 und 3), sind die geltenden Arbeitsschutzmaßnahmen (§§ 8–15) sowie Vorschriften zur Entsorgung kontaminierter Abfälle einzuhalten.
I-7 Zubereitung
Die rezeptur- und defekturmäßige Herstellung erfolgt nach im Voraus erstellten schriftlichen Herstellungsanweisungen, die von einem Apotheker der Krankenhausapotheke zu unterschreiben sind. Bei der rezepturmäßigen Herstellung sind wirkstoffspezifische, bei der defekturmäßigen Herstellung produktspezifische Herstellungsanweisungen zu empfehlen (siehe Appendix 9, beispielhafte Anweisungen). Die Herstellungsanweisung legt eine standardisierte Arbeitsweise zur Sicherstellung der Arzneimittelqualität fest. Abweichungen sind zu dokumentieren.
I-7.1 Herstellungsanweisung
Die Herstellungsanweisungen sollten gemäß §§ 7 Abs. 1a und 8 Abs. 1 ApBetrO mindestens die folgenden Angaben umfassen:
- Geltungsbereich
- Festlegung der Verantwortlichkeiten
- Bezeichnung des Produktes und der Arzneiform und ggf. der Applikationsart (z. B. intrathekal)
- Aufzählung der Ausgangsmaterialien, Primärbehältnisse und Hilfsmittel ggf. mit Lagerort und -bedingungen
- Beschreibung von Maßnahmen zur Vermeidung von Kreuzkontaminationen und Verwechslungen (z. B. Vorbereitung des Arbeitsplatzes)
- Beschreibung der einzelnen Prozessschritte
- Inprozesskontrollen (z. B. Kontrolle der Berechnungen, der Einwaagen/Abmessungen durch eine zweite Person [Vieraugenprinzip] oder durch validierte elektronische Verfahren)
- Kennzeichnung
- Lagerungshinweise (z. B. kühl, lichtgeschützt)
- Haltbarkeit und Aufbrauchfrist
- Angaben zur Verpackung und zum Transport
- Freigabe und Dokumentation
I-7.2 Herstellungsprotokoll
Die Sicherstellung der Arzneimittelqualität erfordert eine umfassende Dokumentation der Herstellung in Form eines Herstellungsprotokolls (siehe Appendix 10, beispielhaftes Protokoll).
Das Herstellungsprotokoll der rezepturmäßigen Herstellung nach § 7 Abs. 1c ApBetrO bzw. das der defekturmäßigen Herstellung nach § 8 Abs. 2 ApBetrO sollte Folgendes beinhalten:
- Name des Patienten und des verschreibenden Arztes (§ 7 ApBetrO) z. B. in Form der Kopie der Verordnung
- Bezeichnung des Arzneimittels (§§ 7, 8 ApBetrO)
- Art und Menge der Ausgangsmaterialien und deren Chargenbezeichnungen (§§ 7, 8 ApBetrO)
- Arzneiform und ggf. Applikationsart (z. B. intrathekal) (§§ 7, 8 ApBetrO)
- Apothekeninterne Herstellungsnummer oder Chargenbezeichnung (§ 8 ApBetrO)
- Herstellungsdatum (§ 8 ApBetrO)
- Lagerungshinweis (§§ 7, 8 ApBetrO)
- Verfalldatum (§ 8 ApBetrO)
- Wägeprotokoll oder Wäge- bzw. Abmessdokumentation mit Vieraugenprinzip (§ 8 ApBetrO)
- Herstellungsparameter und Ergebnis der Inprozesskontrollen (§§ 7, 8 ApBetrO)
- Gesamtausbeute und ggf. die Anzahl der abgeteilten Darreichungsformen (§ 8 ApBetrO)
- Name (§ 7 ApBetrO) oder Unterschrift des Herstellers (§ 8 ApBetrO)
- Ergebnis der organoleptischen Prüfung (§ 7 ApBetrO)
- Freigabe durch den verantwortlichen Apotheker (§§ 7, 8 ApBetrO).
Herstellungsanweisung und Herstellungsprotokoll können in einem Dokument zusammengefasst sein.
I-8 Kennzeichnung
Die rezepturmäßig hergestellten Arzneimittel müssen vor der Abgabe gemäß § 14 ApBetrO, die defekturmäßig hergestellten Arzneimittel nach § 10 AMG gekennzeichnet werden (siehe Appendix 11, beispielhafte Kennzeichnungen). Soweit für das Rezepturarzneimittel ein Fertigarzneimittel als Ausgangsstoff eingesetzt wird, genügt anstelle der Angabe des Wirkstoffs und der sonstigen Bestandteile die Angabe der Bezeichnung des Fertigarzneimittels. Die Kennzeichnung muss in gut lesbarer und in dauerhafter Weise erfolgen. Dazu werden Etiketten mit den erforderlichen Angaben versehen und dauerhaft auf das Primärbehältnis aufgebracht.
I-9 Prüfung
I-9.1 Prüfung bei der rezepturmäßigen Herstellung
Bei einer Rezeptur kann nach § 7 Abs. 2 ApBetrO von einer analytischen Prüfung des Produkts abgesehen werden, sofern die Qualität durch das Herstellungsverfahren, die organoleptische Prüfung des fertig hergestellten Arzneimittels und, soweit vorgesehen, durch die Ergebnisse der Inprozesskontrollen gewährleistet ist.
Als nicht zerstörende Prüfungen sind beispielsweise geeignet:
- Organoleptische Prüfung des Arzneimittels (z. B. Färbung, Klarheit, Partikelfreiheit der Lösung, Füllvolumen)
- Unversehrtheit der Primärbehältnisse und die Verwendung des richtigen Primärbehältnisses
- Überprüfung, ob die eingesetzten Materialien sowie das Etikett mit der Herstellungsanweisung und der ärztlichen Verordnung übereinstimmen
- Prüfung auf korrekte Dokumentation der Herstellung
- Prüfung auf Plausibilität des Gehalts
- Dichtigkeitsprüfung: Kein sichtbarer Austritt von Flüssigkeit aus dem Primärbehältnis, bei flexiblen Primärbehältnissen auch nicht unter Anwendung von Druck
I-9.2 Prüfung bei der defekturmäßigen Herstellung
Die Prüfung bei der defekturmäßigen Herstellung ist risikoadaptiert durchzuführen und sollte die Überprüfung der Herstellungsdokumentation beinhalten. Bis zur Freigabe sind die Produkte unter Quarantäne zu lagern. Es ist eine Prüfanweisung zu erstellen, in der die Art der Probenahme und die Prüfmethode festzulegen sind. Diese sollte die Sterilitätsprüfung mittels Aliquotmethode und automatisierten Nachweismethoden (Schnellnachweis mit automatisierten Blutkulturflaschensystemen) und mindestens 2 Tage Bebrütung umfassen. Auf eine Gehaltsbestimmung kann bei der Verwendung von Fertigarzneimitteln als Ausgangsstoff verzichtet werden und bezüglich des Gehalts eine parametrische Freigabe erfolgen. Die Prüfung und ihre Ergebnisse sind in einem Prüfprotokoll zu dokumentieren. Die Freigabe erfolgt durch den verantwortlichen Apotheker und ist zu dokumentieren.
Prüfanweisung und Prüfprotokoll können in einem Dokument zusammengefasst sein (s. Appendix 12,13 beispielhafte Prüfanweisung und Prüfdokumentation).
Produkte, die nicht den Anforderungen genügen, sind unverzüglich zu separieren.
I-10 Mikrobiologische Validierung und Monitoring der aseptischen Herstellung
Die Qualität der aseptischen Herstellung ist durch geeignete, validierte Verfahren sicherzustellen.
I-10.1 Prozessvalidierung
Die Validierung der Herstellungsprozesse erfolgt durch Simulation mittels Nährmedien (Nährmedienabfüllung). Die Nährmedienabfüllung ist als Eingangsprüfung von jedem Mitarbeiter, der die Tätigkeit in der aseptischen Herstellung aufnimmt, durchzuführen. Die Überprüfung der aseptischen Arbeitsweise jedes Mitarbeiters ist in definierten Abständen durchzuführen, entweder kontinuierlich (häufige Nährmedienabfüllung in geringem Umfang und summative Auswertung) oder diskontinuierlich (mindestens einmal jährlich in größerem Umfang).
Validierungsverfahren
- Die Simulation sollte die Herstellungsprozesse möglichst genau imitieren. Es sollten möglichst komplexe Prozesse (worst case) abgebildet werden. Die äußeren Bedingungen bei der Simulation sollen den bei der Herstellung üblichen Bedingungen entsprechen. Die mikrobiellen Umgebungsbedingungen sollten während der Nährmedienabfüllung mit Sedimentationsplatten und Abklatschplatten kontrolliert werden.
- Für unterschiedlich durchgeführte Prozesse (z. B. manuell, halbautomatisch, automatisch) soll jeweils ein Simulationsverfahren erarbeitet werden, das sich an dem höchsten Kontaminationsrisiko orientiert, und eine Verfahrensanweisung hierzu erstellt werden (s. Appendix 14, beispielhafte Anweisungen).
- Die Simulation sollte am Ende eines Arbeitstages durchgeführt werden.
- Als Flüssignährmedium wird als Universalmedium Sojapepton-Caseinpepton nach Ph. Eur. empfohlen. Nährmedien dürfen maximal 10% verdünnt werden. Alternativ können doppelkonzentrierte Medien verwendet und durch die Prozesssimulation 1:1 verdünnt werden.
- Zur Bebrütungstemperatur und -dauer der abgefüllten Nährmedien sind die Herstellerangaben zu beachten (z. B. bei Sojapepton-Caseinpepton nach Ph. Eur. 7.3 14 Tage bei 20–25°C). Nach der Bebrütung dürfen die Nährmedien keine Trübung aufweisen.
- In einem Nährmedienabfüllvorgang ist eine dem Produktionsumfang angemessene Anzahl von Produktsimulationen zuzubereiten, z. B. 30 Stück oder jeweils 10 Stück an 3 aufeinanderfolgenden Tagen und pro Mitarbeiter. Anschließend muss mindestens einmal jährlich ein Nährmedienabfüllvorgang kontaminationsfrei durchgeführt werden.
- Die Validierung ist erfolgreich absolviert, wenn in keinem der abgefüllten Nährmedien eine mikrobielle Kontamination nachgewiesen wurde.
- Eine festgestellte Kontamination bei der Eingangs- oder Revalidierung erfordert eine erneute Eingangsvalidierung.
- Für eine festgestellte Kontamination ist eine Keimdifferenzierung zu empfehlen.
- Die durchgeführten Nährmedienabfüllungen sowie deren Ergebnisse sollten übersichtlich dokumentiert werden.
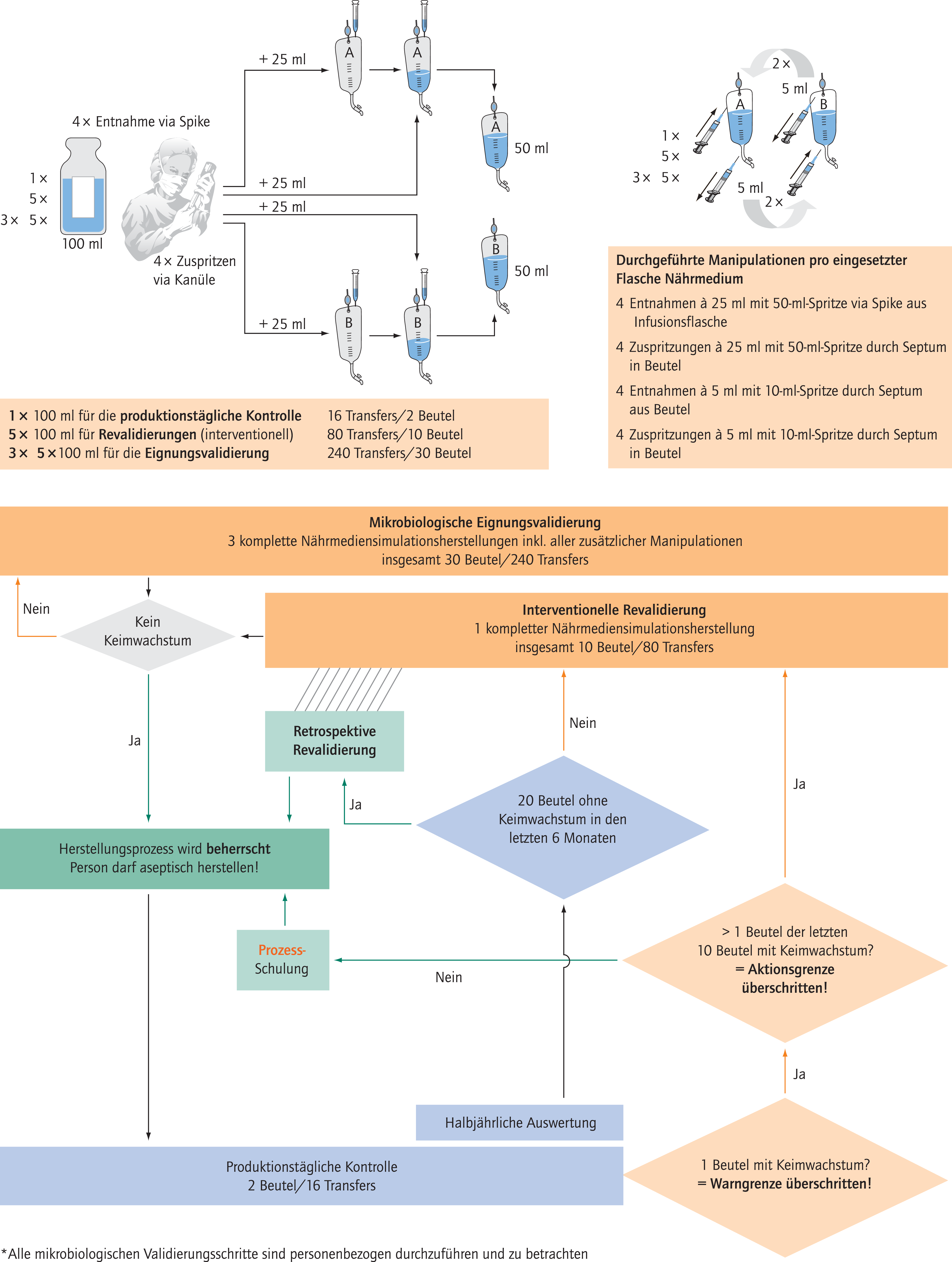
Appendix 14, Beispiel 1. Anweisungen für die Nährmediensimulation.
I-10.2 Prozesskontrolle
Zur Prozesskontrolle können am Ende eines Produktionstages Dummys, Anbrüche oder Rückgaben auf Sterilität geprüft werden. Dies erfolgt mittels automatisierten Schnellnachweismethoden (z. B. Blutkulturflaschensysteme) und Zugabe eines Aliquots. Alternativ können zusätzliche Nährmedienabfüllungen in einem jährlichen Stichprobenumfang von 0,4 mal Wurzel aus der jährlich hergestellten Zahl von applikationsfertigen Parenteralia durchgeführt werden.
I-10.3 Umgebungskontrollen
Die Umgebungsbedingungen sollten durch geeignete Kontrollen der Luft, kritischer Oberflächen und des Personals anhand von Partikel- und Keimzahlbestimmungen während der Herstellung überprüft werden. Sedimentationsplatten und aktive Luftkeimsammler sind für das Monitoring der luftgetragenen mikrobiellen Kontamination geeignet. Zur Kontrolle der mikrobiologischen Qualität kritischer Oberflächen und des Personals sind Abklatschplatten geeignet. Die Mitarbeiter, die die Umgebungskontrollen durchführen, sollten darin geschult sein.
Untersuchungsmethoden
Untersuchungen der Luft
- Passive Luftkeimsammlung: Sedimentationsplatten (Ø 90 mm, Expositionszeit der Arbeitsdauer angepasst, max. 4 Stunden)
- Aktive Luftkeimsammlung: Methode der Wahl im reinen Bereich ist die Luftkeimsammlung mittels Impaktionsverfahren. Empfehlenswert ist ein Luftkeimsammler mit hohem Luftdurchfluss (100 l/min), zu messendes Luftvolumen mindestens 1 m³ pro Messpunkt in Reinraumklasse A
- Partikel: Für die Partikelzählung im reinen Bereich sind Partikelzählgeräte mit hohem Luftdurchfluss (100 l/min) geeignet. Zu messendes Luftvolumen in Reinraumklasse A/B 1 m³ Luft pro Messpunkt oder nach EN-ISO 14644-1:2010 für die Klassen B, C, D:
- im Minimum jedoch mindestens 2 Liter bzw. 1 Minute Probenahmezeit pro Messpunkt
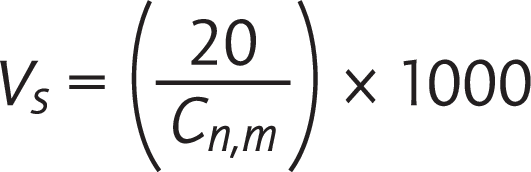
Vs: Mindesteinzelprobenvolumen an einem Probenahmeort, angegeben in Liter; cn,m: Klassengrenze (Partikelzahl je Kubikmeter) für die größte betrachtete Partikelgröße der entsprechenden Klasse; 20: Die bestimmte Partikelanzahl, welche gezählt werden könnte, wenn die Partikelkonzentration sich an der Klassengrenze befindet.
Untersuchungen kritischer Oberflächen
- Abklatschplatten (Ø 55 mm)
Untersuchungen des Personals (Finger, Bereichskleidung)
- Abklatschplatten (Ø 55 mm)
Als Nährmedium für alle Platten eignet sich Agarmedium mit Caseinpepton und Sojapepton. Die Abklatschplatten sollten Zusätze („Enthemmer“) enthalten, um antimikrobiell wirkende Stoffe zu neutralisieren. Zur Bebrütungstemperatur und -dauer der Platten sind die Herstellerangaben zu beachten (z. B. bei Sojapepton-Caseinpepton nach Ph. Eur. 7.1 3–5 Tage bei 30–35°C). Die Auswertung erfolgt durch Auszählung der koloniebildenden Einheiten je Platte. Für eine festgestellte Kontamination ist eine Keimdifferenzierung zu empfehlen.
Anzahl und Lokalisation der Messpunkte
Passive Luftkeimsammlung
- Anzahl: repräsentativ für den Raum bzw. Bereich (z. B. LAF, Schleuse)
- Messpunkte: an potentiell kritischen Stellen (z. B. LAF, Arbeitsflächen, Tischschleusen). Die Auswahl der Messpunkte ist schriftlich zu begründen.
Aktive Luftkeimsammlung
- Anzahl: repräsentativ für den Raum (z. B. die Hälfte der errechneten Anzahl Messpunkte der Partikelzählung)
- Messpunkte: 1,20 m über dem Boden, an potentiell kritischen Stellen (z. B. an Türen, vor den LAFs, an häufigen Laufwegen). Die Auswahl der Messpunkte ist schriftlich zu begründen.
Partikel
- Anzahl: √m2 der Raumfläche des Reinraums aufgerundet auf eine ganze Zahl (z. B. 3 bei 9 m², 4 bei 12 m² Raumfläche)
- Messpunkte: 1,20 m über dem Boden, an potentiell kritischen Stellen (z. B. an Türen, vor den LAFs, an häufigen Laufwegen). Die Auswahl der Messpunkte ist schriftlich zu begründen.
Abklatschtest von Oberflächen
- Anzahl: repräsentativ für den Raum bzw. Bereich
- Messpunkte: an potentiell kritischen Stellen (z. B. LAF, Arbeitsflächen, Tischschleusen). Die Auswahl der Messpunkte ist schriftlich zu begründen.
- Anpressdruck: 500 g
- Dauer: 10 sec
Abklatschtest Personal
- Anzahl: beide Hände, jeweils 5 Fingerkuppen
- Messpunkte: an potentiell kritischen Stellen (z. B. Fingerkuppen, Bereichskleidung)
Basismonitoring
Die Umgebungsbedingungen werden durch ein Basismonitoring qualifiziert. Dazu wird eine angemessene Zahl repräsentativer und potentiell kritischer Messpunkte definiert und über einen begrenzten Zeitraum, z. B. innerhalb von 14 Tagen, täglich ein Messwert (Methoden s. oben) genommen (in Summe mindestens 10 Messwerte pro Messpunkt).
Aus den ermittelten Werten werden Basiswerte (arithmetische Mittelwerte) gebildet, anhand derer die Reinraumklasse klassifiziert wird.
Routinemonitoring
Anhand der Ergebnisse des Basismonitorings wird ein Prüfplan für ein Routinemonitoring-Programm erarbeitet, wobei folgende Punkte definiert werden müssen:
- Messpunkte: Reduktion auf die beim Basismonitoring ermittelten repräsentativen und kritischen Messpunkte
- Methoden: s. o.
- Hinweise zu Prüffrequenzen sind z. B. dem PIC/S PE 10 Leitfaden zu entnehmen (siehe Appendix 15, Vorschlag zu Probenfrequenzen)
- Warn- und Aktionsgrenzen: Ausgehend von den beim Basismonitoring ermittelten Basiswerten (B) müssen Warn- (WG) und Aktionsgrenzen (AG) festgelegt werden, deren Überschreiten erhöhte Aufmerksamkeit bzw. ein Einschreiten erfordert (beispielhaft siehe Appendix 16). Zu beachten ist, dass die Warn- und Aktionsgrenzen sich innerhalb der ermittelten Reinraumklasse befinden sollten.
- Maßnahmen bei Überschreiten der Warn- und Aktionsgrenzen
Appendix 15. Vorschlag für die Probenfrequenz beim Routineumgebungsmonitoring.
Unter der Maßgabe der täglichen Prozesskontrolle sind alternative Konzepte zur PIC/S PE10-Empfehlung möglich. Die unterschiedlichen Angaben zu den möglichen Probefrequenzen wurden bei der Erstellung des Vorschlags berücksichtigt.
|
PIC/S PE10 |
Vorschlag |
|
|
Partikelzählung Kritische Zone |
Vierteljährlich |
Halbjährlich |
|
Partikelzählung Umgebung |
– |
Halbjährlich |
|
Passive LKZ Kritische Zone |
Immer |
Wöchentlich |
|
Passive LKZ Umgebung |
Wöchentlich |
Wöchentlich |
|
Aktive LKZ Kritische Zone |
Vierteljährlich |
Vierteljährlich |
|
Aktive LKZ Umgebung |
Vierteljährlich |
Vierteljährlich |
|
Oberflächen Kritische Zone |
Wöchentlich |
Wöchentlich |
|
Oberflächen Umgebung |
Monatlich |
Wöchentlich |
|
Finger |
Nach jeder Arbeitsperiode |
2xwöchentlich |
LKZ: Luftkeimzahl
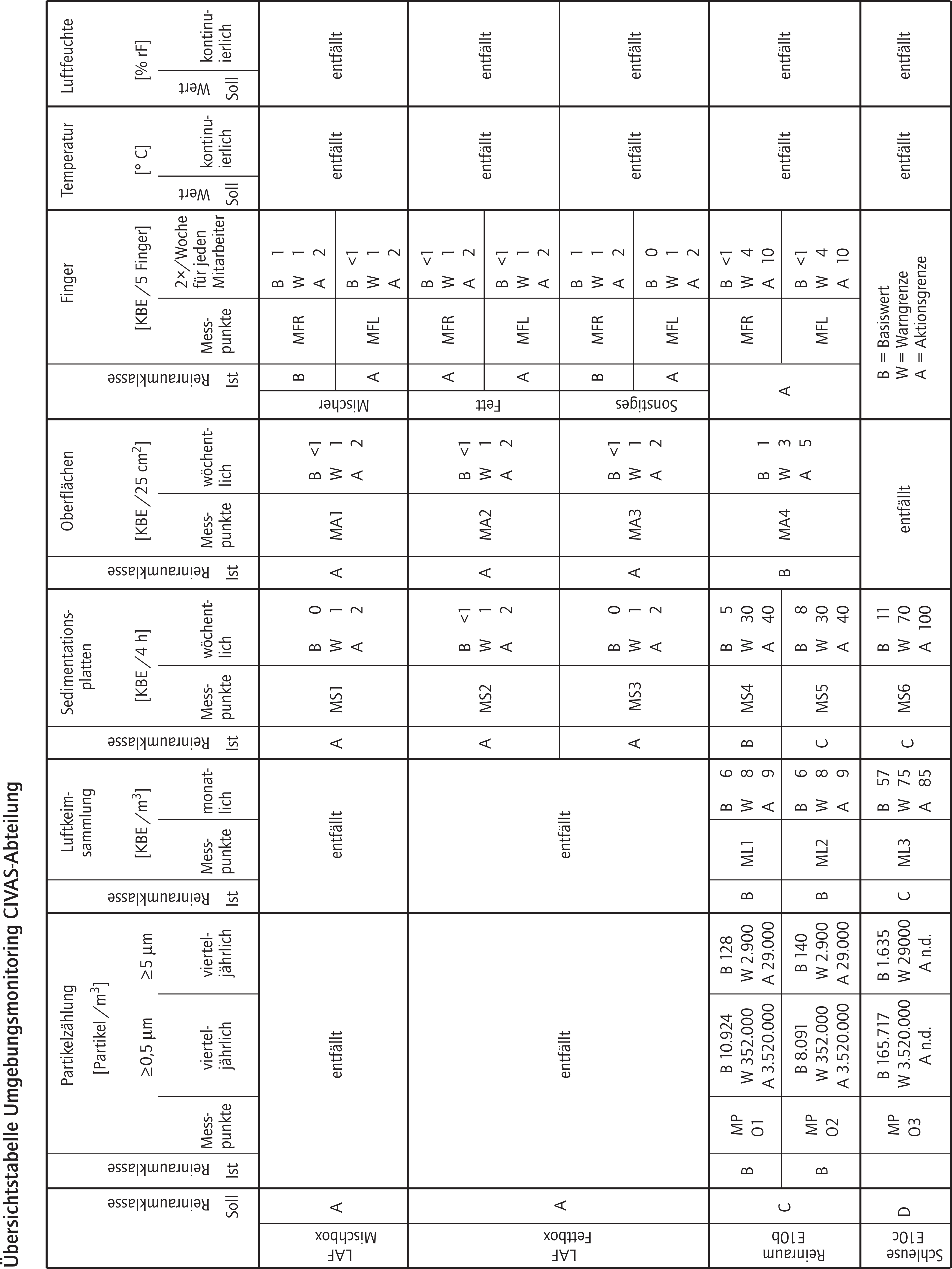
Appendix 16. Beispiel für Basiswerte und Warn- und Aktionsgrenzen für das Routinemonitoringprogramm
Die Ergebnisse der Umgebungskontrollen sind zu dokumentieren und regelmäßig zu überprüfen, um Abweichungen im Sinne einer unerwünschten Erhöhung der Keim- oder Partikelbelastung rechtzeitig festzustellen und geeignete Maßnahmen einleiten zu können. Es empfiehlt sich das Führen von Trendanalysen (Beispiel siehe Appendix 17).
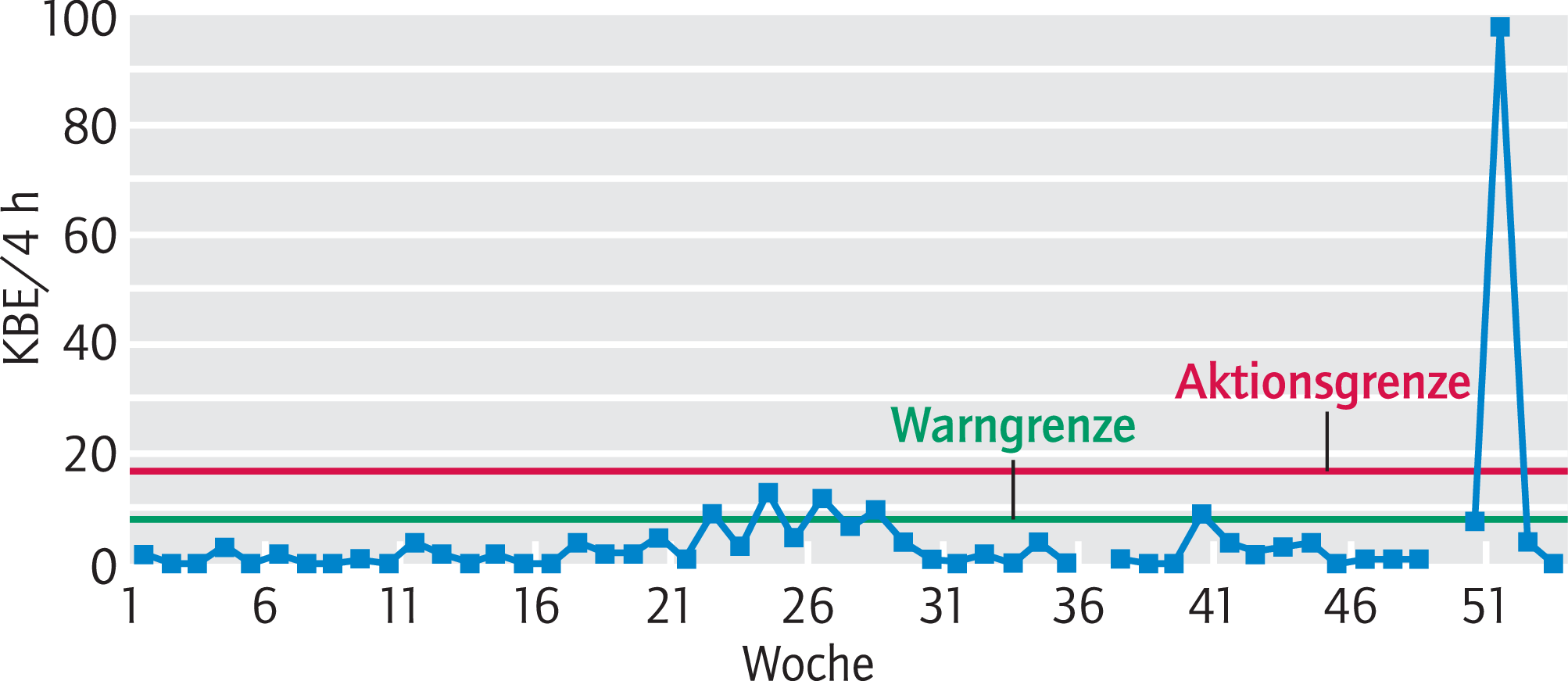
Appendix 17. Beispiel für eine Trendanalyse.
I-11 Dokumentation
Bei Verwendung einer Herstellungssoftware kann die Dokumentation entsprechend angepasst werden. Gemäß § 22 ApBetrO müssen alle Dokumente mindestens fünf Jahre archiviert werden.
I-12 Lagerung/Transport und Verwendbarkeitsfristen
Die hergestellten applikationsfertigen Parenteralia werden in keimarme Verpackungen einzelverpackt und verschlossen. Die Verpackung sollte so gestaltet sein, dass sie einen Originalitätsverschluss und Auslaufschutz darstellt. Eine Kühllagerung ist zu bevorzugen, wenn es die physikalisch-chemische Stabilität zulässt. Die Festlegung der Verwendbarkeitsfristen liegt in der Verantwortung des zuständigen Apothekers. Die Bewertung der physikalisch-chemischen Stabilität der Zubereitung erfolgt auf Grundlage eigener Untersuchungen oder auf Grundlage publizierter experimenteller Untersuchungen. Die Übertragbarkeit der Literaturdaten ist umso eher möglich, je ähnlicher die untersuchten Zubereitungen (beispielsweise hinsichtlich Primärbehältnis, Trägerlösung, Konzentration) den in Frage stehenden Zubereitungen sind. Die Verwendbarkeitsfrist ist in der Herstellungsanweisung festzulegen. Der Transport soll unter Beachtung der Stabilität und bei CMR-Arzneimitteln mit den notwendigen Schutzmaßnahmen erfolgen.
I-13 Literatur
Verordnung über den Betrieb von Apotheken, Apothekenbetriebsordnung (ApBetrO) Juni 2012. www.bgbl.de/Xaver/start.xav?startbk=Bundesanzeiger_BGBl&start=//*[@attr_id=‘bgbl112s1254.pdf‘]
Pharmaceutical Inspection Cooperation Scheme (PIC/S) PE 010-3 Guide to Good Practices for the preparation of medicinal products in healthcare establishments. October 2008. www.picscheme.org/pdf/23_pe0103-revisedgppguide.pdf
Ph. Eur. 7.1 (incl. Preparationes Pharmaceuticae, Draft Monograph)
Council of Europe Resolution CM/ResAP (2011; Januar 2011. https://wcd.coe.int/ViewDoc.jsp?id=1734101&Site=CM
USP 35/NF 30, Mai 2011 <797> Pharmaceutical Compounding – Sterile Preparations
Kramer A, Assadian O (Hrsg.). Wallhäußers Praxis der Sterilisation, Desinfektion, Antiseptik und Konservierung. Stuttgart: Thieme Verlag, 2008.
Baumann L, Maurer J. Retrospektive Revalidierung. Krankenhauspharmazie 2003;24:471–9.
Krämer I. Zytostatikaherstellung in der Apotheke. Pharmazie in unserer Zeit 2010;39:280–7.
Arbeitsstättenverordnung vom 12. August 2004 (BGBl. I S. 2179), zuletzt durch Artikel 4 der Verordnung vom 19. Juli 2010 (BGBl. I S. 960) geändert.

Appendix 2, Beispiel 2. Plausibilitätsprüfung von CMR-Arzneimitteln zur parenteralen Anwendung
Krankenhauspharmazie 2013; 34(02)