Le Hang Pelzl, Heidelberg, Judith Thiesen, Mainz, Ina-Maria Klut, Dresden
GCP-konforme Mitarbeit an klinischen Prüfungen ohne Herstellungserlaubnis – Rekonstitution klinischer Prüfpräparate
1. Zweckbestimmung und Geltungsbereich
Die Leitlinie beschreibt die Standards für eine GCP-(Good Clinical Practice-)konforme Mitarbeit der Krankenhausapotheke an klinischen Prüfungen im Rahmen des üblichen Apothekenbetriebes. Sie soll als Orientierung für Art, Umfang und Qualität zu erbringender Dienstleistungen und zu erfassender Daten sowie als Grundlage für die Erstellung interner Verfahrens- und Arbeitsanweisungen dienen. Diese Leitlinie gilt für klinische Prüfungen mit Arzneimitteln, die keiner Herstellungserlaubnis gemäß § 13 Arzneimittelgesetz (AMG) bedürfen, unabhängig davon, ob als Sponsor ein pharmazeutischer Unternehmer, eine akademische Institution oder eine Fachgesellschaft auftritt. Die Mitarbeit der Krankenhausapotheke mit über die Rekonstitution gemäß § 4 (31) AMG hinausgehenden Herstellungstätigkeiten wird Gegenstand weiterer Leitlinien sein.
2. Regulatorische Anforderungen
Die Sicherstellung der ordnungsgemäßen Qualität der von der Krankenhausapotheke erbrachten Leistungen erfolgt unter Beachtung der allgemein anerkannten pharmazeutischen Regeln. Insbesondere sind in der jeweils aktuell gültigen Fassung zu berücksichtigen:
- Gesetz über den Verkehr mit Arzneimitteln (AMG): §§ 4, 13, 14, 40, 41, 42, 42a, 67
- Verordnung über die Anwendung der guten klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Arzneimitteln zur Anwendung am Menschen (GCP-Verordnung, GCP-V)
- Leitlinie zur guten klinischen Praxis (ICH-GCP) (CPMP/ICH/135/95)
- Richtlinie 2001/20/EG des Europäischen Parlaments und des Rates zur Angleichung der Rechts- und Verwaltungsvorschriften der Mitgliedstaaten über die Anwendung der guten klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Humanarzneimitteln (EU-GCP)
- Apothekenbetriebsordnung (ApBetrO): §§ 3, 6
Die speziellen Aspekte von „Good Manufacturing Practice“ (GMP), deren Einhaltung die GCP-V ausdrücklich fordert, sind im Rahmen des üblichen Apothekenbetriebes nicht verpflichtend. Eine daran orientierte Vorgehensweise ist jedoch empfehlenswert. Geltungsbereiche anderer Gesetze sind im Einzelfall nicht ausgeschlossen und müssen entsprechend berücksichtigt werden.
3. Zuständigkeiten
Für die ordnungsgemäße Mitarbeit an der klinischen Prüfung tragen der zuständige Apotheker und der Apothekenleiter die Verantwortung.
Der für die klinische Prüfung zuständige Apotheker und sein Stellvertreter sind namentlich zu benennen. Sie fungieren als Hauptansprechpartner gegenüber Sponsor, Contract Research Organisation (CRO) und Prüfzentrum. Die fachliche Eignung ist in der Regel durch einen Lebenslauf nachzuweisen.
Gemäß § 3 ApBetrO dürfen pharmazeutische Tätigkeiten, zum Beispiel Rekonstituieren, ausschließlich durch pharmazeutisches Personal ausgeführt werden. Tätigkeiten wie Warenlogistik und Dokumentation dürfen hingegen auch von nicht pharmazeutischem Personal ausgeführt werden.
4. Qualitätsstandard für die GCP-konforme Mitarbeit an klinischen Prüfungen ohne Herstellungserlaubnis
Siehe Abbildung 1.
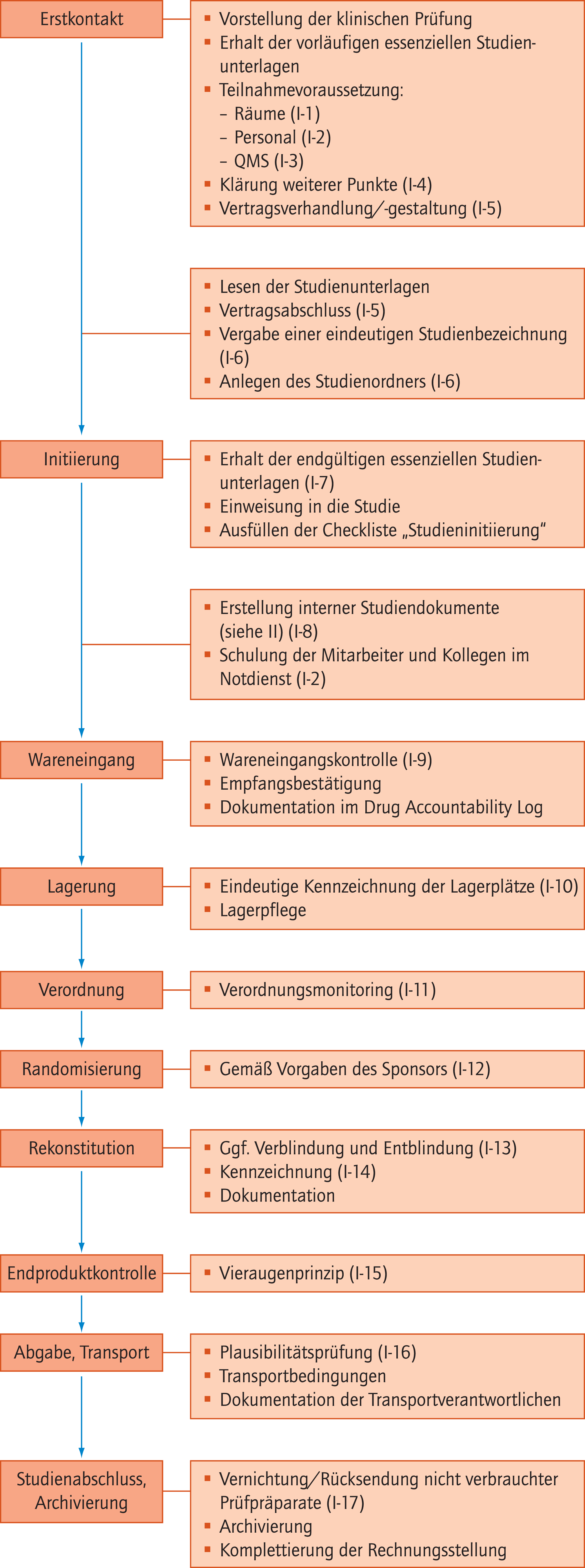
Abb. 1. Fließdiagramm zur Darstellung des Qualitätsstandards für die GCP-(Good Clinical Practice-)konforme Mitarbeit der Krankenhausapotheke an klinischen Prüfungen ohne Herstellungserlaubnis
Anhang
I Ausführungen zum Fließdiagramm
I-1 Räume
Anforderungen an Lagerorte
Klinische Prüfpräparate müssen getrennt von regulären Arzneimittelbeständen gelagert werden. Wenn möglich, sind Lagerorte in einem separaten, abschließbaren Raum oder Schrank bzw. Kühl-/Tiefkühlschrank einzurichten. Der Lagerort muss gekennzeichnet und vor dem Zugriff Unbefugter geschützt sein. Die einzelnen Lagerplätze der verschiedenen Prüfpräparate sind mit eindeutiger Studienbezeichnung zu versehen. Ebenso sollte ein separater Lagerort für verfallene oder aus anderen Gründen nicht verwendbare Prüfpräparate eingerichtet werden.
Klinische Prüfpräparate sind so zu lagern, dass die vom Sponsor vorgegebenen Bedingungen eingehalten werden. Über die Einhaltung der Lagerbedingungen ist ein Nachweis zu führen. Der Nachweis sollte durch tägliches Monitoring der Umgebungsbedingungen (Temperatur, ggf. Luftfeuchte) mit im Messbereich kalibrierten Geräten (z. B. Minimum-Maximum-Thermometer, Digitalthermometer mit Min-Max-Aufzeichnung, zentrales Temperaturüberwachungssystem; Kalibrierungszertifikat mit gültiger Laufzeit) erbracht werden. Die erhaltenen Daten (aktuelle Temperatur/Minimum/Maximum, ggf. Luftfeuchte) sind unter Berücksichtigung der jeweiligen Grenzwerte zu beachten und zu dokumentieren. Temperaturüberwachungssysteme mit Alarmmeldung haben den Vorteil, dass bei Temperaturüber- oder -unterschreitungen eine unverzügliche Reaktion erfolgen kann. Ist das Einrichten eines Alarmsystems nicht möglich, muss mindestens die Aufzeichnung von Abweichungen sichergestellt werden.
Abweichungen sind durch den zuständigen Apotheker schriftlich zu bewerten. Im Einzelfall kann die Abweichung von den vorgeschriebenen Lagerbedingungen die Kontaktaufnahme zu Sponsor/CRO zur Klärung der weiteren Vorgehensweise erforderlich machen. In dieser Zeit müssen die betroffenen Prüfpräparate mit eindeutiger Studienbezeichnung getrennt gelagert werden und dürfen bis zur Klärung nicht verwendet werden.
Anforderungen an Herstellungsräume
Die für die patientenindividuelle Rekonstitution genutzten Räumlichkeiten und die Ausrüstung müssen für diese Tätigkeit geeignet, in funktionstüchtigem Zustand und gewartet sein. Reinigung und Desinfektion müssen gründlich und so ausgeführt sein, dass Kreuzkontaminationen und andere qualitätsgefährdende Einflüsse ausgeschlossen werden können.
I-2 Personal
Die Apotheke muss für die Mitarbeit an klinischen Prüfungen über Personal in ausreichender Zahl und mit der erforderlichen Qualifikation und Erfahrung verfügen. Der zuständige Apotheker hat die betreffenden Mitarbeiter vor der Arbeitsaufnahme und dann fortlaufend, zum Beispiel einmal jährlich, zu relevanten Themen, zum Beispiel GCP und Personal- bzw. Raumhygiene, zu schulen und dies zu dokumentieren. Der Schulungserfolg ist durch geeignete Maßnahmen zu überprüfen. Die Schulungsnachweise sind gemäß GCP mindestens 10 Jahre aufzubewahren.
Darüber hinaus müssen vor Beginn jeder klinischen Prüfung die betreffenden Mitarbeiter in die studienspezifischen Details eingewiesen werden. Hierbei sind auch, soweit zutreffend, Kollegen des Spätdienstes und der Rufbereitschaft zu berücksichtigen. Die Einweisungen sind zu dokumentieren und von den Beteiligten mit Datum und Signum zu versehen. Einweisungsnachweise sind in der Apotheke im Studienordner zu archivieren.
Bei apothekenrelevanten Prüfplanänderungen sind die Mitarbeiter erneut einzuweisen.
Die Aufgaben und Verantwortlichkeiten aller Studienbeteiligten werden im Delegation of Authority/Responsibility Log geregelt. Hier werden in der Regel auch die Namenskürzel der Studienbeteiligten festgelegt.
I-3 Qualitätsmanagementsystem
Aus dem etablierten Qualitätsmanagementsystem (QM-System) sollten dem Sponsor/der CRO Beschreibungen allgemeiner Prozesse zur Verfügung gestellt werden können. In einem Organigramm sollte ersichtlich sein, welche Personen in welcher Funktion für klinische Prüfungen zuständig sind.
Im QM-System sollten Verfahrens- und Arbeitsanweisungen für folgende Prozesse enthalten sein:
- essenzielle Studienunterlagen
- Vertragswesen
- Warenlogistik, Lagerung und Bestandsführung klinischer Prüfpräparate
- Rekonstitution klinischer Prüfpräparate und Dokumentation
- Randomisierung/Verblindung/Notfallentblindung klinischer Prüfpräparate
- Vernichtung klinischer Prüfpräparate
- Studienabschluss und Archivierung von Studiendokumenten
I-4 Vorbereitungen einer klinischen Prüfung
Im Vorfeld einer klinischen Prüfung müssen alle besonderen Anforderungen eruiert und organisiert werden. Hierzu kann zum Beispiel der Transport klinischer Prüfpräparate mit sehr kurzer Verwendbarkeit oder unter nachgewiesenen und dokumentierten Transportbedingungen (z.B. 2 bis 8°C) an das Prüfzentrum gehören. Ebenso muss geklärt werden, ob die Mitarbeit der Krankenhausapotheke außerhalb der regulären Dienstzeit erforderlich ist.
Die gegebenenfalls erforderliche Bereitstellung von Hilfsmitteln zur Rekonstitution oder Applikation von klinischen Prüfpräparaten sowie von Begleitmedikation durch die Apotheke ist im Vorfeld zu besprechen und festzulegen.
I-5 Vertrag
Auf der Basis der im Vorfeld ermittelten bzw. dem Prüfplan entnommenen Angaben ist eine Leistungsvereinbarung zwischen Sponsor/Auftraggeber und Apotheke/Auftragnehmer abzuschließen, in der die Verantwortlichkeiten klar geregelt sind. Bestandteil des Vertrags sind auch die Vereinbarungen zur Vergütung der Leistungen.
Zu berücksichtigende Aspekte sind zum Beispiel:
- Warenlogistik
- Rekonstitution klinischer Prüfpräparate und gegebenenfalls der Begleitmedikation
- Dokumentation
- Leistungen außerhalb der regulären Dienstzeit
- Beschaffung von Hilfsmitteln, Begleitmedikation u.a.
- Monitoring- und Auditbesuche
- Vernichtung klinischer Prüfpräparate
- gegebenenfalls Overhead-Kosten der Verwaltung
Eine Einmalpauschale (setup-fee) zur Abdeckung der Initiierungsleistungen sollte immer Bestandteil des Apothekenvertrags sein.
I-6 Interne Studienbezeichnung/Studienordner der Apotheke (Pharmacy File)
Der klinischen Prüfung muss eine eindeutige Bezeichnung (Studienname und Studiennummer) zugeordnet werden, die mit der Bezeichnung im Prüfzentrum übereinstimmt. Dies gewährleistet im Verlauf der klinischen Prüfung die Identifizierung und reduziert bei der Mitarbeit an vielen Projekten Missverständnisse oder die Verwechslung von Prüfpräparaten. Alle vorliegenden Dokumente sind in einem Ordner zusammenzufassen. Aus Gründen der Vertraulichkeit darf nur befugtes Personal Zugriff darauf haben.
I-7 Initiierung
Vor dem Start einer klinischen Prüfung findet ein Initiierungsbesuch (initiation visit) durch den Sponsor/die CRO im Prüfzentrum statt. Dieser übergibt der Apotheke spätestens jetzt einen Studienordner (Pharmacy File) mit den finalen studienrelevanten Unterlagen, zum Beispiel Prüfplan, Pharmacy Manual, Investigators Brochure und Formblätter zur Dokumentation. Bislang der Apotheke vorliegende Dokumente sind diesem Ordner zuzuführen (siehe I-6).
Weiterhin erfolgt die Besprechung des Prüfplans, des Prüfpräparates und der speziellen Aufgaben der Apotheke. Eine Checkliste mit wichtigen Punkten ist hierbei hilfreich (siehe Checkliste für die Studieninitiierung). Die Übergabe von Studienunterlagen sowie die Initiierung sind schriftlich zu dokumentieren und durch Unterschrift zu bestätigen. Die Initiierung sollte erst stattfinden, wenn der Vertrag, zumindest vom Auftraggeber unterschrieben, vorliegt.
I-8 Erstellung interner Studiendokumente
Es ist empfehlenswert, eine spezifische Arbeitsanweisung für jede klinische Prüfung zu erstellen. Diese sollte alle wichtigen Angaben prüfplangerecht zusammenfassen und sowohl zur Einweisung der Mitarbeiter als auch zur Information im Verlauf der klinischen Prüfung geeignet sein (siehe I-2).
Formblätter zur Dokumentation werden in der Regel vom Sponsor/der CRO zur Verfügung gestellt. Sind keine oder ungeeignete Formblätter vorhanden, sollten diese von der Apotheke erstellt werden. Eine Auflistung essenzieller Unterlagen zur GCP-konformen Dokumentation findet sich unter Punkt II.
I-9 Warenlogistik
Die Logistik der Prüfpräparate und alle damit im Zusammenhang stehenden Tätigkeiten sind durch benanntes Personal auszuführen.
Bei Wareneingang muss die Lieferung auf Richtigkeit/Übereinstimmung mit den Angaben des Lieferscheins, Unversehrtheit, gegebenenfalls spezielle Transportbedingungen und die Vollständigkeit von Unterlagen, zum Beispiel Analysen- bzw. Freigabezertifikat des Sponsors, geprüft werden. Studienspezifische Anforderungen, zum Beispiel das Rücksenden von Temperaturschreibern, sind ebenfalls zu berücksichtigen. Der Empfang der Prüfpräparate ist in der vom Sponsor vorgesehenen Weise, zum Beispiel schriftlich per Fax oder per Post bzw. elektronisch per IVRS/IWRS (interactive voice and web response system) zu bestätigen. Treten Unstimmigkeiten auf, so ist der Sponsor/die CRO zu kontaktieren und das Prüfpräparat bis zur Klärung unter Quarantäne zu lagern. Warenzu- und -abgänge sind auf dem entsprechenden Formblatt (Drug Accountability Log) zu dokumentieren. Zu den Angaben gehören in der Regel:
- Bezeichnung der klinischen Prüfung
- gegebenenfalls Zentrumsnummer
- Bezeichnung des klinischen Prüfpräparats
- Datum der Warenbewegung
- Menge der Zu- bzw. Abgänge
- gegebenenfalls Patientenidentifikation (bei patientenbezogener Abgabe)
- gegebenenfalls Medikations-, Flaschen- oder Packungsnummer
- Chargenbezeichnung
- Verfalldatum
- resultierender Bestand
- Datum/Signum des Dokumentierenden
I-10 Lagerung
Prüfpräparate müssen gemäß den Vorgaben des Sponsors gelagert werden (siehe I-1). Die Umgebungsbedingungen, die Verfalldaten, der Bestand (inkl. Übereinstimmung mit dokumentiertem Bestand) sowie der ordnungsgemäße Zustand der Prüfpräparate müssen regelmäßig kontrolliert werden.
Diskrepanzen zwischen dem dokumentierten und dem tatsächlichen Lagerbestand müssen zeitnah geklärt werden. Sollte sich die Diskrepanz nicht aufklären lassen, muss diese zusammen mit einer Erklärung schriftlich festgehalten werden. Korrekturen in Unterlagen haben generell mit Datum und Signum des Korrigierenden, gegebenenfalls unter Angabe eines Grunds, zu erfolgen.
Nachbestellungen klinischer Prüfpräparate müssen rechtzeitig und in Relation zum aktuellen Screening- bzw. Rekrutierungsstand erfolgen. Ein „Stockout“ muss unbedingt vermieden werden.
I-11 Verordnung
Die Verordnung klinischer Prüfpräparate muss unter Angabe der internen Studienbezeichnung (siehe I-6) erfolgen.
Vor der Herstellung ist eine Prüfung auf Plausibilität durch das Vieraugenprinzip (Verordnungsmonitoring) durchzuführen. Bei Verordnungsirrtümern/Abweichungen vom Prüfplan oder Unklarheiten muss Rücksprache mit dem Prüfarzt gehalten werden.
I-12 Randomisierung
Bei der Randomisierung sind die Vorgaben des Sponsors zu befolgen. Nur autorisierte Mitarbeiter dürfen Randomisierungen durchführen. Die erfolgte Randomisierung ist unter Angabe des Datums und Signum der ausführenden Person zu dokumentieren. Es müssen Maßnahmen getroffen und deren Einhaltung sichergestellt werden, die ein versehentliches Entblinden verhindern.
I-13 Rekonstitution
Die Rekonstitution klinischer Prüfpräparate hat gemäß den gültigen nationalen Bestimmungen und gemäß Prüfplan zu erfolgen. Die Herstellung ist zu dokumentieren (Herstellungsdokumentation bzw. Drug Preparation Log).
Zu den Angaben gehören:
- Bezeichnung der klinischen Prüfung
- Bezeichnung/Stärke des klinischen Prüfpräparats
- Patientenidentifikation
- Chargenbezeichnung/Verfalldatum des verwendeten klinischen Prüfpräparates
- Datum und gegebenenfalls Uhrzeit der Rekonstitution
- Signum der zubereitenden Person
- Signum des zuständigen Apothekers
- gegebenenfalls aufgetretene Abweichungen oder Zwischenfälle, so dass eine lückenlose Rückverfolgung des Ablaufs möglich ist
Zusätzlich muss jeder Verbrauch klinischer Prüfpräparate im Drug Accountability Log dokumentiert werden. Es sollte sich mit dem Sponsor/der CRO möglichst darauf verständigt werden, dass leere und angestochene Gefäße klinischer Prüfpräparate unmittelbar nach der Rekonstitution (ggf. dokumentiert) vernichtet werden. Dies gilt insbesondere für CMR-Arzneimittel (CMR: kanzerogen, mutagen, reproduktionstoxisch). Bei doppelblinden Studien ist entsprechend den Vorgaben des Sponsors auf eine vollständige Verblindung der zubereiteten Prüfpräparate zu achten, zum Beispiel durch Lichtschutzbeutel und ein gefärbtes Infusionssystem bei Parenteralia.
Notfallentblindung
Bei verblindeten Prüfpräparaten hat der Sponsor vor Beginn der klinischen Prüfung ein Verfahren festzulegen, das die unverzügliche und sichere Entblindung gewährleistet. Es ist sicherzustellen, dass die Entblindung nur so weit offengelegt wird, wie dies erforderlich ist. Notfallentblindungen sind nur durch autorisiertes Personal und in der Regel erst nach Genehmigung des Sponsors mithilfe von Notfallentblindungskuverts, Randomisationscodes, IVRS/IWRS oder anderen Systemen vorzunehmen. Jede Entblindung ist in geeigneter Form zu dokumentieren.
I-14 Kennzeichnung
Die Angaben auf den Etiketten der rekonstituierten Zubereitung sollten sich an den Vorgaben des § 5 GCP-V orientieren. Ausgewählte Inhalte können auch auf einem Begleitdokument, zum Beispiel der Patienteninformation, aufgeführt werden.
Um bei einem doppelblinden Konzept sowohl die Verblindung als auch die Patientensicherheit zu gewährleisten, sollten die in Frage kommenden Wirkstoffe und ihre Dosierungen auf dem Etikett aufgeführt sein.
I-15 Endproduktkontrolle
Vor Abgabe einer Zubereitung zur klinischen Prüfung muss eine dokumentierte Endproduktkontrolle durch autorisiertes, pharmazeutisches Personal durchgeführt werden (Vieraugenprinzip).
I-16 Abgabe und Transport
Sofern Prüfpräparate ohne Rekonstitution abgegeben werden, hat vor der Abgabe ebenfalls eine Prüfung auf Plausibilität analog I-11 zu erfolgen. Die studienspezifischen Transportbedingungen, zum Beispiel Kühlkette, spezielle Transportbehälter, sind zu beachten. Bei CMR-Arzneimitteln sind die einschlägigen Vorgaben, zum Beispiel verschließbare, bruchsichere Behältnisse, zu berücksichtigen. Bei Bedarf ist ein Transportverantwortlicher zu benennen und entsprechend zu schulen. Der Transport wird gegebenenfalls nach Anweisung des Sponsors dokumentiert.
I-17 Studienabschluss und Archivierung
Der Abschluss einer klinischen Prüfung erfolgt mit dem Abschlussbesuch (close-out visit). Hierbei wird die Dokumentation im Studienordner der Apotheke finalisiert und, falls noch nicht erfolgt, die Vernichtung oder Rücksendung verbrauchter und nicht verbrauchter Prüfpräparate (Dokumentation im Drug Destruction Log) durchgeführt. Nach Beendigung der Studie sind die Unterlagen entsprechend GCP-V für mindestens 10 Jahre bzw. entsprechend den Vorgaben des Sponsors zu archivieren. Die Komplettierung der vertragsgemäßen Rechnungsstellung ist zu überprüfen, und es ist gegebenenfalls die Schlussrechnung zu stellen.
I-18 Monitoring Visits, Audits, Inspektionen
Monitoring Visits durch den Sponsor/CRO dienen in der laufenden klinischen Prüfung der Überprüfung der vertrags- bzw. prüfplangemäßen Leistungserbringung der Apotheke. Im Mittelpunkt steht der ordnungsgemäße Umgang mit klinischen Prüfpräparaten, insbesondere Lagerung/Nachweis der Lagerbedingungen, Rekonstitution, Bestandsführung und Dokumentation dieser Tätigkeiten. Es sollte sich mit dem Monitor darauf verständigt werden, dass Monitor-Besuche nur nach vorheriger Terminvereinbarung stattfinden.
Audits oder Inspektionen durch den Sponsor oder Behörden dienen der Überprüfung der Mitarbeit der Apotheke an klinischen Prüfungen. Im Mittelpunkt stehen in der Regel die Überprüfung von Einrichtungen, Personal, Unterlagen und Dokumentation hinsichtlich der Übereinstimmung von Planung und realer Durchführung sowie der Einhaltung rechtlicher Grundlagen. Die Teilnahme der Apotheke an Audits oder Inspektionen ist obligat.
II Liste essenzieller Dokumentationsunterlagen
- Checkliste für die Studieninitiierung
- Delegation of Authority/Responsibility Log
- Namentliche Nennung der Studiendurchführenden, Festlegung der Verantwortlichkeiten
- Drug Accountability Log: Bilanzierung des Prüfpräparates
- Drug Destruction Log: Dokumentation der Vernichtung des Prüfpräparates
- Drug Preparation Log: Dokumentation der Rekonstitution des Prüfpräparates
- Schulungs-/Einweisungsnachweis
- Studienspezifische Arbeitsanweisung
- Temperature Log: Dokumentation der Lagertemperatur
- Verordnungsformular (mit Angabe der eindeutigen Studienbezeichnung)
Dr. Le Hang Pelzl, Apotheke des Universitätsklinikums Heidelberg, Im Neuenheimer Feld 670, 69120 Heidelberg, E-Mail: lehang.pelzl@med.uni-heidelberg.de,
Dr. Judith Thiesen, Apotheke der Universitätsmedizin Mainz, Langenbeckstr. 1, 55131 Mainz, Dr. Ina-Maria Klut, Apotheke des Universitätsklinikums Dresden, Fetscherstraße 74, 01307 Dresden
Pharmacy services in clinical trials exempt from manufacturing licence according to the GCP-guideline – reconstitution of investigational medicinal products
Hospital pharmacies have become more and more important cooperation partners for sponsors of clinical trials. The most requested service is the reconstitution of investigational medicinal products (IMP) according to § 4(31) of the German Medicines Law (German: AMG) including related services such as logistics, storage, documentation and destruction of IMPs. The guideline presented here describes standards for the performance of clinical trials according to the GCP-guideline and the Ordinance on the Operation of Pharmacies (German: ApBetrO). The guideline serves as a framework to evaluate the type, scope and quality of the provided pharmacy services as well as the collected data. Moreover, it may serve as a basis for the compilation of internal operating and working instructions. The guideline is applicable for clinical trials on medicines which are exempt from a manufacturing licence. There is no difference whether a pharmaceutical company, an academic institution or a professional association acts as sponsor. A guideline for pharmacy services other than reconstitution according to § 4 (31) German Medicines Law should be the subject of further guidelines.
Key words: Hospital pharmacy, guideline, clinical trial, reconstitution
Krankenhauspharmazie 2012; 33(02)